|
|
 |
™
|
| News, April 17, 2005 |
|
| Thank you for visiting our page. We are working to improve it. New cases coming soon. |
|
Granulomatous Dermatitis/Vasculitis
3/31/2003
This case is currently under treatment at the Dept. of Dermatology at UTMB. The clinical history was contributed by Dr. Renee R. Snyder, a second year dermatology resident. If you can help with the diagnosis or treatment please let me know by writing your comments in the "Contact Us" section of this webpage.
The patient is a 61 y/o male who was first seen in UTMB in October 2002. He had one year history of confluent, extensive, painful erythematous plaques, some of which appeared annular. (Clinical figures).
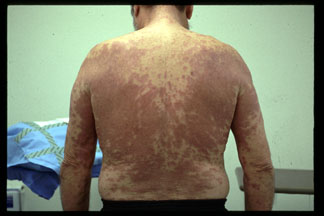
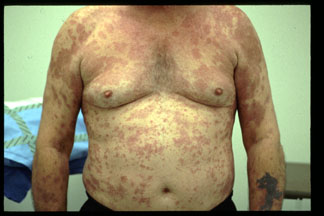
A previous biopsy by the referring dermatologist had been read as "granuloma annulare" and the working diagnosis was disseminated GA. Lab work-up was all WNL including C-Anca,P-Anca, RF and ANA. There was increased C-reactive protein of 11.9 (0-5).
Patient has been treated with Dapsone X2 months, Prednisone, and narrow band UVB without response. Presently he is on Prednisone 10 mg/d, and Cyclophosphamyde, without clear improvement so far.
New biopsies of skin were obtained. (See histologic pictures).
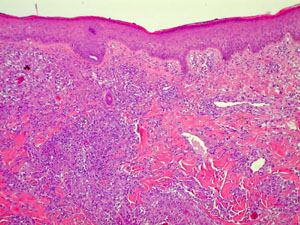
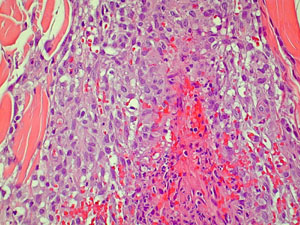
The histology showed a prominent interstitial histiocytic infiltrate, predominantly in the upper dermis, with vascular involvement, i.e. granulomatous vasculitis/dermatitis. IP stains for B and T-cells were mostly negative, while CD68 (histiocytes) was markedly positive.
The dermal infiltrate is too extensive to make the diagnosis of interstitial granuloma annulare; furthermore, no mucin is present.
Two other entities come to mind with these findings. First is "interstitial granulomatous dermatitis" (IGD) which presents with annular plaques, often with raised border, located mainly on upper trunk or shoulders. IGD has been associated with RA, collagen vascular disease or drug reactions. Our patient clinical findings do not fit completely with IGD. Histologically IGD is characterized by a interstitial histiocytic infiltrate in the deep dermis and subcutaneous tissue, without vascular involvement and with abundant eosinophils.
The other possibility has been described under multiple headings including "cutaneous extravascular necrotizing granuloma", "Churg-Strauss granuloma", and "neutrophilic and palisaded granulomatous dermatitis". This entity presents clinically with papules, and sometimes annular plaques; fingers and elbows are common sites. The condition has been associated with sistemic vasculitis (including allergic granulomatosis, Wegener's and polyarteritis), connective tissue diseases, lymphoproliferative disiases, and other entities such as SBE, chronic active hepatitis and ulcerative colitis.
The histology in our case is most consistent with this last entity although no underlying condition has been identified so far in this patient.
Do you have ideas regarding this case?
|
|
|




